
Managing Impurities During Clinical Trials

 Impurity control is a critical aspect of any pharmaceutical development process, but regulatory guidance tends to be vague where limited use during clinical trials is concerned.
Impurity control is a critical aspect of any pharmaceutical development process, but regulatory guidance tends to be vague where limited use during clinical trials is concerned.
According to AstraZeneca’s Andrew Teasdale: “Permissible limits for impurities during clinical development has always been a grey area. Existing guidelines, specifically ICH Q3A, only formally apply to marketed products. Factors such as duration of clinical trials have never been effectively addressed.”
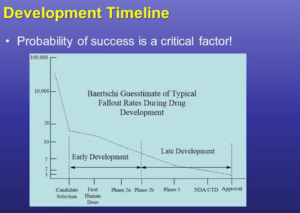
Since impurity levels can have a significant impact on patient safety, it’s important for you, as the drug sponsor, to lay out a strategy for discovering, measuring, identifying and qualifying them early on – even in the pre-clinical phase.
This approach minimizes your risk and can potentially save significant time and money. That’s because early identification of impurities enables you to understand the mechanism of impurity formation — and take steps to correct it — at the earliest possible point in development. If necessary, you can find alternate suppliers of raw materials, or take steps to remove or reduce the impurities (for example, by modifying synthetic processes or isolations), while minimizing disruption at later stages.
Each product’s particular impurity control strategy will be influenced by a variety of factors, including phase of development, toxicology profile, process capability, indication, route of administration, dose, and duration of the trial.
Types of Pharmaceutical Impurities
A pharmaceutical impurity is any component that is not the chemical entity defined as the drug substance or an excipient in the drug product. Impurities fall into three categories:
- Organic impurities — These can result from the starting materials, intermediates, APIs or degradation products, be process-related impurities which arise during manufacturing, or occur during storage.
- Inorganic impurities —These include reagents, ligands, catalysts, inorganic salts, heavy metals/elemental impurities or other residual metals. Filter aids and charcoals are other materials that also fall into this category.
- Residual solvents —These are organic or inorganic liquids used during various steps of the manufacturing process. Because these generally have a known level of toxicity, the selection of appropriate controls is easily accomplished.
The levels of impurities which exist in your drug substance need to be qualified with biological safety studies.
The Role of ICH Guidelines
The management of related substance organic impurities is defined by quality guidelines issued by the International Conference on Harmonisation (ICH). Standards for commercial new drug substances are defined by ICH Guidance Q3A. While this is considered the “guiding star” for development chemists, clinical candidates have more impurity flexibility since the clinical trial patient only takes the drug for a limited time. That said, early-phase substances must still be justified against an established toxicology profile. For mutagenic impurities ICH M7 covers both clinical and commercial situations.
 Pharma companies often take different approaches to the ICH guidelines on impurities. Some want their specifications to be as strict as the commercial guidelines from Day One, while others are less stringent in the early stages. While both approaches have pros and cons, the guidelines influence specifications in either case. It’s always worthwhile to “listen to the footsteps” of the commercial requirements and develop your product with the end in mind.
Pharma companies often take different approaches to the ICH guidelines on impurities. Some want their specifications to be as strict as the commercial guidelines from Day One, while others are less stringent in the early stages. While both approaches have pros and cons, the guidelines influence specifications in either case. It’s always worthwhile to “listen to the footsteps” of the commercial requirements and develop your product with the end in mind.
Realistically, however, successfully developing a drug, completing clinical trials and entering the commercial market is a high-risk – and high cost – undertaking. The choice to put off developing comprehensive impurity control strategies is frequently driven by cost, especially in earlier clinical (or pre-clinical) stages.
ICH Thresholds
ICH Q3A has a very helpful chart that defines which organic impurities meet the thresholds for reporting, identification, and qualification. These thresholds may motivate you to do additional development work to not only reduce the number of qualified impurities, but also to reduce future work. If you’re able to get under a threshold, you must be confident that you can remain there despite any variances, which will inevitably arise in your process.
Again, identifying impurities as early as possible is critical. It’s bad enough if a new impurity slows your project by rising to identification or qualification level in late development. The impact will be much worse if this happens during the commercial phase. To minimize risk, you should meet ICH guidelines by clinical Phase 2B.

Information in the above table is referenced in document ICH Q3A. The full document can be viewed here.
One of the most commonly referenced numbers from this chart is the threshold of 0.1% for identification. While the threshold is intended for drug product but often applied to drug substance. That’s because any impurity greater than this level may require extensive work to identify, purify and qualify in toxicology studies.
ICH also has well-defined management standards for residual solvents (ICH Q3C) and elemental impurities (ICH Q3D). We recommend that you try to avoid using class 1 or 2 solvents during development.
Given their ability to damage DNA, mutagenic or potentially genotoxic impurities (PGIs) warrant much more concern – with even the smallest amounts posing a risk. Controlling genotoxic impurities (GTIs) is essential for every substance entering clinical trials. Chemists can often look at alerting structures in the molecule. There are also some software products which can predict if an impurity is mutagenic.
TTC: Threshold of Toxicological Concern
The level of regulatory concern is roughly a 1,000-fold difference for a mutagen/PGI versus a typical organic impurity. Because genotoxic impurities may be present at extremely low levels (ppm), analytical methods must be very sensitive, requiring the use of techniques such as mass spectrometry. ICH Guideline M7 Mutagenic Impurities covers both clinical trial and commercial products (below).

Information from the above table is referenced in document ICH M7. The full document can be viewed here.
 For cancer drugs which are often themselves mutagenic, guidance ICH S9 gives you more flexibility on impurities – and often higher thresholds can be set. The rationale is that the cancer therapeutics used to treat cases are themselves potentially dangerous compounds, and any inherent impurities may not be as harmful as the therapeutic compound itself.
For cancer drugs which are often themselves mutagenic, guidance ICH S9 gives you more flexibility on impurities – and often higher thresholds can be set. The rationale is that the cancer therapeutics used to treat cases are themselves potentially dangerous compounds, and any inherent impurities may not be as harmful as the therapeutic compound itself.
Minimize Risk by Partnering with a CDMO
Working with a CDMO is one of the best ways you can streamline the management and identification of impurities. This benefits you in three ways:
- Input from process chemists can help narrow down possibilities based on the synthesis.
- Your CDMO has access to starting materials, intermediates, mother liquors, and by-products, which significantly assists the process of impurity identification.
- If needed, your CDMO also has the ability to isolate or synthesize quantities of the impurity for reference or qualification studies.
Regis has both the process and analytical knowledge to support your management of impurities during clinical trials. Read more about our approach to identifying and controlling drug impurities.
Video Resources
Speeding Drug Development through Impurity Control Strategies

Selected Case Studies and Impurity Strategies for Drug Substances by Paul Wrezel, Ph.D.

Analytical Package, CMC and Impurities
