
Analytical Strategies from Early Development to Validation: Part One

The Role of Analytical Method Development and Validation
Drug development is complex and requires great care to ensure that the products we offer our patients are safe and effective. Consequently, drug development is highly regulated to ensure the appropriate controls are applied for all new chemical entities (NCEs), whether they are marketed products or still in development. While current guidelines on quality do exist, such as the recently published DRAFT guideline Q14s published by International Conference on Harmonization (ICH) in March 2022, current guidelines are more detailed and prescriptive for commercial drug products and substances. Less guidance exists for NCEs that are in the clinical phases of development.
During drug development, analytical chemists play an important role as they must strategically create test methods and control strategies that guide the process development effort with developing, optimizing, and scaling up for routine manufacturing. Additionally, analytical chemists are responsible for guiding formulation scientists who are in the midst of developing drug product dosage forms. These processes and procedures are necessary to ensure the identity, strength, quality, purity, and potency of each drug substance and drug product manufactured for human use.
Regis Technologies recently recorded an online presentation which details the analytical strategies that support CMC drug development from the preclinical phase to commercial production, which you may view on our website. In this blog, we summarize the first part of that presentation.
Regulatory Guidance for Drug Development
As mentioned earlier, the drug development process is highly regulated to ensure patient safety. Analytical procedures exist and are necessary to ensure the identity, strength, quality, purity, and potency of drug substances used by humans.
The ICH has published detailed guidelines and current regulatory guidelines apply to both commercial drug substances and drug products (ex: ICH, FDA, EMA). You may access the ICH guidance database at any time by visiting www.ich.org.
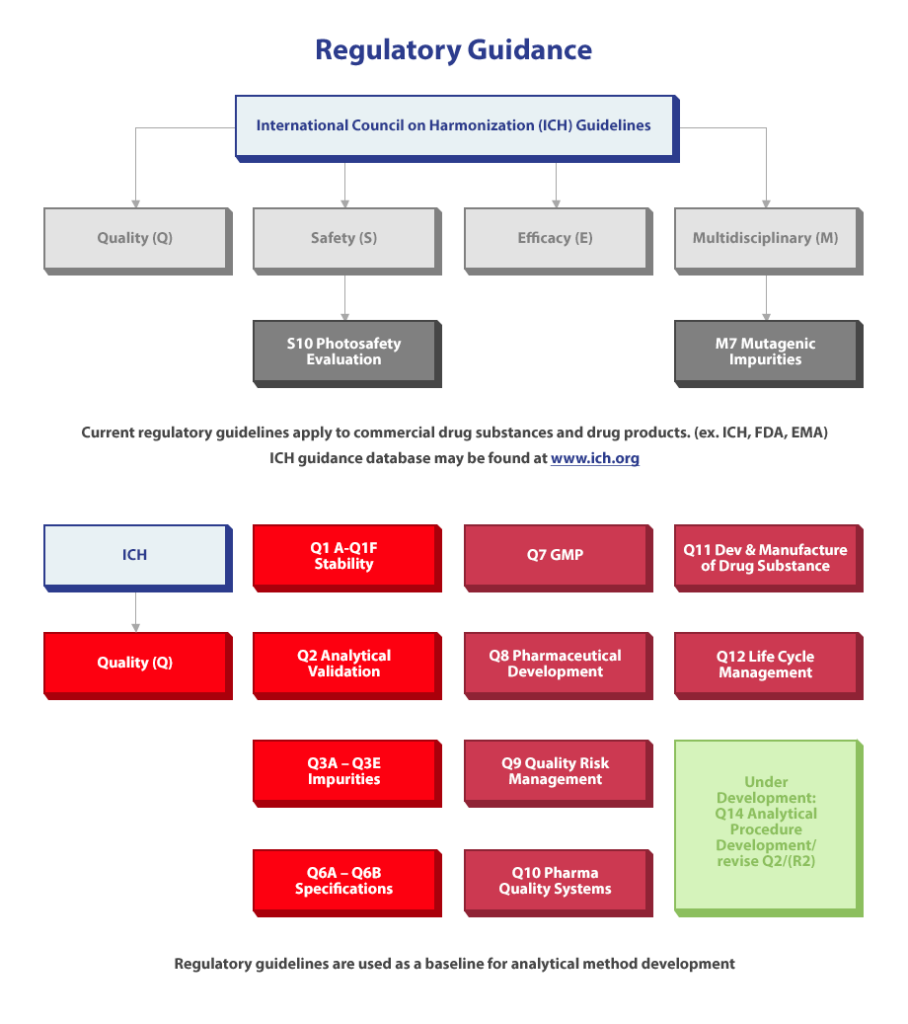
Regulatory guidelines are used as a baseline for analytical development.
The Purpose of Analytical Methods
Analytical scientists develop test methods to guide process chemists, guide formulation scientists, test final drug substance API, and determine fit-for-purpose, types, and techniques.
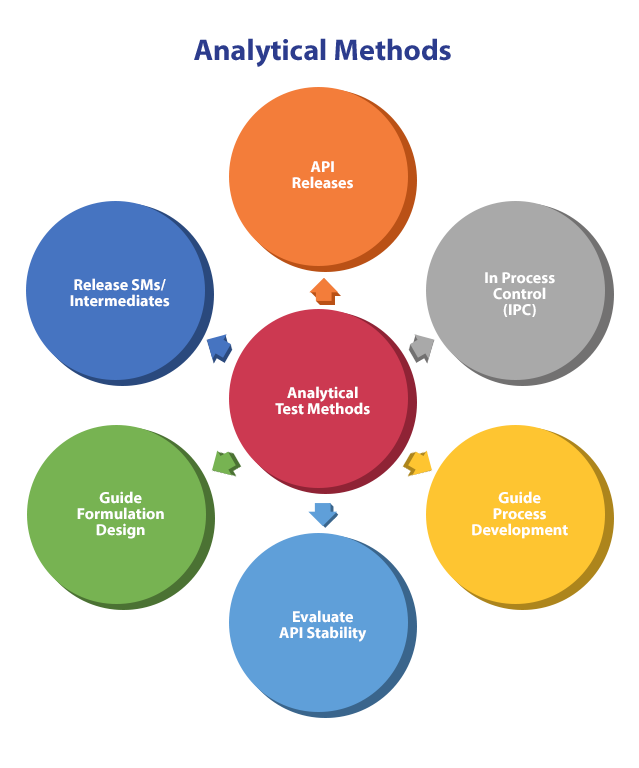
The Role of API Characterization Tests
API Characterization Tests are conducted to ensure the identity, strength, quality, purity, and potency of each drug substance and drug product manufactured for human use. These tests are designed to examine:
- Identification
- Content
- Organic Impurities
- Inorganic Impurities
- Volatile Content
- Counterion Content
- Potential Genotoxic Impurities
- Particle Size
- Solid State Characterization
Analytical Development for Each Phase of Clinical Development

Preclinical: The goal of the preclinical phase is to assign a purity to drug substances used for toxicology dosing studies and qualify impurities present in API in animal studies. Some typical analytical procedures include purity, TGA, Micro ROI, and Alternative Techniques.
Phase 1: The goal of phase 1 is to assign purity to drug substances for First In Human (FIH) or safety studies and to evaluate impurity levels. Additionally:
- Early phase 1 methods must be scientifically sound, considering that at the early phases, there is a limited patient population and only short-term studies.
- Impurities must be limited to those in toxicology batches tested in preclinical animals, profile and level comparison.
- Typical methods for API include:
- Qualified (Scientifically Sound) Test Methods
- Appearance
- Identification (NMR/IR)
- Purity (%AUC) and Impurities (%AUC)
- Residual Solvents
- Water Content
- ROI
- Chiral or Conterion (if needed)
- Elemental Impurities
Phase 2A/2B: In this phase, the goal is to assign purity to drug substances for a larger patient population and to evaluate impurity levels.
To do this, chemists may or often modify the process. Then, they will evaluate analytical test methods with the new process to isolate and identify key impurities. Impurities are limited to those in toxicology batches tested in animals and qualified impurity levels. If a new impurity appears, it must be qualified, which takes time and money, and may delay your program. A genotoxic assessment of impurities should also be performed during this phase.
Typical methods for API include:
- Validated Test Methods
- Appearance
- Identification (NMR/IR)
- Purity (%AUC) or Assay (%w/w) and Related Substances
- Residual Solvents
- Water Content
- ROI
- Chiral or Counterion (If Needed)
- Elemental Impurities
These Methods should be validated by the end of phase 2.
Phase 3/Registration: The goal during phase 3 is to have a well-characterized API for pivotal clinical studies, as well as to control impurity levels to meet proposed commercial targets.
In this phase, commercial scale chemical processes are locked prior to pivotal clinical batches, typical analytical development includes additional impurity studies, and GMP batches must have impurities at or below those qualified in toxicology studies (no new impurities at or >0.10%).
Typical methods for API include:
- Appearance
- Identification (NMR/IR)
- Purity (%AUC) or Assay (%w/w) and Related Substances
- Residual Solvents (NMT ICH Limits)
- Water Content
- ROI
- Chiral or Counterion (If Needed)
- Elemental Impurities
Test methods must be validated before phase 3.
Commercial: The goal for this phase is to ensure all API critical quality attributes meet regulatory standards (ex: ICH).
At this phase, starting materials, intermediates, and API should all be well characterized. Additionally, impurity profiles should be understood and well controlled, analytical control strategy should be well defined, all test methods should be validated, and the manufacturing process should be validated.
Conclusion
We hope you have found this overview of Analytical Method Development to be not only educational, but helpful. In the next few weeks, we will publish Part Two of this article which will explore Analytical Method Validation.